
El día 2 de Abril del 2013 acude un varón de 63 años por clínica hemorrágica (epistaxis, hematuria, gingivorragia).
Antecedentes personales: sin interés, no alergias conocidas, no tto habitual.
Enfermedad actual: refiere síntomas gripales desde hace 2 semanas consistentes en sensación distérmica, no termometrada, tos seca y molestias osteomusculares. Ha tomado Azitromicina durante 3 días y un compuesto homeopático de metales (cobre, plata y oro). Desde hace 4 días refiere epistaxis, gingivorragias, hematuria sin coágulos y dolor lumbar que el paciente relaciona con la tos.
Exploración general: PA 123/69 mmHg. Fc 72 lpm. Tª 36ºC. SatO2 95%. Consciente y orientado, bien hidratado y perfundido, eupneico, coloración de piel y mucosas normal, ampollas hemorrágicas en mucosa oral: yugal y debajo de la lengua; petequias en el paladar. Restos de sangre en ambas fosas nasales sin sangrado activo. PETEQUIAS en ambas piernas, cara y espalda. Resto anodino.
Pruebas complementarias: analítica:
– Bioquímica: Bilirrubina, Creatinina, Urea, Iones (Cl, Na, K y Ca), Glucosa, Proteínas totales, TSH, LDH, GPT, GOT, GGT, FA: normales
– Hemograma: Serie roja y blanca: normales. PLAQUETAS: 2 *10e3/uL (normal: 140-400). Plaquetas inmaduras: 54% (normal: 1,1-6,1 )
– Frotis de sangre periférica: sin alteraciones morfológicas significativas. Trombopenia confirmada. No esquistocitos. Algún linfocito hiperbasófilo de aspecto activado.
– Tº de Protrombina (INR) y TTPA: normales.
– Queda pendiente: Proteinograma. Inmunoglobulinas. Serología de: Hepatitis (B y C), VIH, Citomegalovirus, Herpes simplex (1 y 2), Parvovirus B19, Virus de Epstein Barr.
* TROMBOPENIA severa (2.000 plaquetas/mm3) asociada a clínica hemorrágica.
.
Causas de trombopenia:
0) Pseudotrombopenia por EDTA. (agregados plaquetarios)
1) Centrales (producción deficiente): aplasia/hipoplasia de la médula ósea, transtornos de la maduración, procesos malignos…
2) Por secuestro: hiperesplenismo, hepatopatía, hipotermia.
3) Periféricas (destrucción acelerada):
a) Inmune:
– Inmune primaria: PTI
– Inmune secundaria: Fármacos, Enfermedades autoinmunes (LES…), VIH, Infecciones…
b) No inmune (microangiopáticas): PTT, SHU, CID, Sindrome de HEELP….
.
Cuestiones:
a) En nuestro caso cuál es el diagnóstico más probable? -> PTI: PÚRPURA TROMBOPÉNICA IDIOPÁTICA (Trombocitopenia inmune primaria)
b) Tratamiento? -> Corticoides, Inmunoglobulinas IV… AntiD (Rh) en pacientes Rh+ no esplenectomizados, Transfusión de plaquetas (duran muy pocos días) …. Rituximab … Esplenectomía… Agonistas del receptor de la trombopoyetina: Romiplostim y Eltrombopag... Inmunodepresores/inmunomoduladores (azatioprina, ciclosporina, danazol, dapsona, micofenolato)…
c) Pregunta un familiar del paciente cuál es su pronóstico ??? -> aproximadamente el 10 por ciento de los pacientes adultos con PTI no responde adecuadamente a las diferentes combinaciones terapéuticas (Glucocorticoides, Inmunoglobulinas IV, Rituximab, Esplenectomía, etc) = PTI crónica refractaria.
.
EVOLUCIÓN: nuestro paciente ha sido tratado en principio con Corticoides a dosis de 1 mg/Kg y posteriormente a dosis más altas + Inmunoglobulinas IV (3 días) + Transfusión de un pool de plaquetas. Pese a dicho tratamiento hoy día 9 sigue con plaquetas muy bajas (1.000/mm3) por lo que se inicia tto con ROMIPLOSTIN (agonista del receptor de la trombopoyetina).



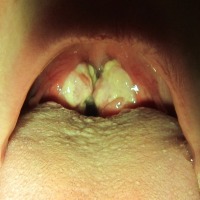




Muy bueno, saludos